浅谈美国保健品市场的法规要求及应对策略
□ 陶鑫 Martin Hahn 美国霍金路伟律师事务所华盛顿办公室
美国保健品市场的发展较为成熟稳定,中国企业要想进入美国保健品市场,如何应对法规要求是重中之重。保健品在美国通常被称为膳食补充剂或饮食补充剂(dietary supplements)。膳食补充剂虽然不能像药品一样标明具体适应症,但可以标示声称对人体功能结构具有保健作用。自美国《膳食补充剂健康及教育法案》(The Dietary Supplement Health and Education Act of 1994简称“DSHEA法案”)于1994年颁布以来,美国的保健品市场迎来了迅猛的发展。据估计,美国保健品的销售额在2017年将达到361亿美金。随着美国的“婴儿潮”一代(特指美国二战后生育高峰)逐渐步入老年,他们对维系健康生活方式的渴求和强劲的消费能力都毋庸质疑地会给美国的保健品市场提供更多新的发展契机。
随着美国保健品消费群体对中国医食同源等东方健康哲学的逐渐接受,以及近年来对天然、非化学合成产品的推崇,源自中草药或其他基于天然植物中活性成分开发的产品在美国市场有很强的商业潜力。中国作为保健品及原料的生产大国,如何让好的产品成功地进入美国市场,了解相关的法规要求是重中之重。下面仅就保健品及原料进入美国市场的基本法规要求及应对策略做一简析。
DSHEA法案如何定义“膳食补充剂”
作为保健品,合法在美销售的最基本要求是明确产品符合DSEHA法案中膳食补充剂的定义。DSHEA法案将膳食补充剂归为食品的一个特殊种类,膳食补充剂是对正常饮食的补充且含有一种或多种膳食补充成分(dietary ingredient)。DSHEA法案要求膳食补充剂必须是口服的,其服用形式可以包括丸剂、胶囊、粉、软胶囊、片剂、散剂、或溶液等各种形式。尽管口服的要求很好理解,但FDA近年来在给企业的警告信(Warning Letter)中多次以此为法律依据判定口腔喷雾和外用涂膏等产品不符合保健品的定义。比如2012年,在FDA给一家名为“Breathable Foods”的公司发的警告信中就判定其销售的可吸入式的咖啡因产品因并非符合口服要求所以不能作为膳食补充剂销售。
膳食补充成分被DSHEA法案定义为包括维生素、矿物质、药草或其他植物类、氨基酸、以及其他可以添加到饮食中的物质。膳食补充剂还可以是任何前述物质的浓缩物、代谢物、组成物、萃取物或是以上所列物的组合物。尤为值得一提的是,FDA对化学合成成分是否符合膳食补充成分的定义有着详细的规定。在2016年颁布的FDA指南文件(草案)《 膳食补充剂:新膳食补充成分的申报和相关问题》中指出,在实验室和工厂生产出来的与天然植物活性成分完全相同的化学合成产品一般不符合膳食补充成分的定义。但是,对于在食品中已经有过应用的,包括像香兰素和肉桂酸等,通常由化学合成生产并用于调味的食品成分,即使是化学合成,也可以被认定为符合DSHEA法案中膳食补充成分的定义。对于希望能够在美销售化学合成的保健品原料企业来说,如果将原料首先应用于食品,便可一定程度上避免一些因为不符合DSHEA法案中膳食补充成分的定义而不能将合成品应用于保健品中的情况。
“首先上市”条款及应对策略
DSHEA法案中最容易被忽视的,但同时对企业前期商业可行性调研也最为重要的条款是所谓的“首先上市”条款。在该条款之下,如果特定的膳食补充成分在美国已经被作为药品的活性成分获批,或者还未获批,但已经作为药品在研而且经过了大量公开的临床研究,那么此成分除了下面列举的情况之外将不能作为膳食补充剂销售。具体来讲,对于已经被批准为新药的某特定成分只有在下面两种情况之下可以作为膳食补充剂销售:其一是FDA将其批准为新药前已经作为膳食补充剂合法销售;其二是FDA制定特别的法规授权其作为膳食补充剂销售。“首先上市”条款的制定是为了保护制药行业在新药研发上的巨大投入不会受到保健品行业的不当侵犯。同理,对于作为药物在研,并经过大量的临床研究且已经公开的成分,也只有在类似的两种情况下可以作为膳食补充剂销售:在大量的临床研究开始以前已经作为保健品销售,或得到FDA的特别法规授权。
FDA曾运用“首先上市”条款在1997年判定Pharmanex Inc.公司的一种名为 “Cholestin”产品不能作为膳食补充剂销售。该产品含有与当时的新药洛伐他汀(lovastatin)同样的活性化学成分。因为含有此种活性成分的膳食补充剂在洛伐他汀被FDA作为新药被批准之前并没有在市场上销售,FDA引用“首先上市”条款判定该产品不符合膳食补充剂的定义并禁止其按照原配方继续销售。对于生产研发保健品原料的企业来说,前期调研应包括研究该特定的成分是否已经被FDA批准为新药的活性成分,是否作为药物在研和其相关的研究进度,以及此原料用于膳食补充剂在美国销售的历史。这些信息将帮助企业自我审核其产品是否有触发FDA引用“首先上市”条款的风险。
新的膳食补充成分审批
新的膳食补充成分在DSHEA法案中被定义为“在1994年10月15号以前没有在美国销售过的膳食补充成分。”在DSHEA法案下,除非被认定是新的膳食补充成分,这样保健品的上市才不需要经过FDA对原料的审批。保健品的上市前原料审核有3种情况:一是企业进行自我评估后发现膳食补充剂不含有任何新的膳食补充成分;二是企业评估后发现新的膳食补充成分但与该成分化学上相同的原料已经应用于食品中;三是企业评估发现新的膳食补充成分且该成分未应用于食品中。3种情况中只有最后一种需要向FDA递交新膳食补充成分的申报(New Dietary Ingredient Notification)。
新膳食补充成分的申报必须在产品上市前75天递交给FDA。申报时应提供该成分在预定的使用条件之下可以被认为是安全的科学证据。在FDA给企业拒绝申请的回函中最常提到的问题是安全数据的不完整。尽管对安全数据的要求不难理解,FDA还是在已经公开的审核中拒绝了大多数新膳食补充成分的申报。FDA建议新成分申报中的安全数据最好是来源于对原料的膳食补充安全性专门进行研究的实验。但是,在人们看到的大批公开的新成分申报中,企业引用的安全数据却多是在原料的效用研发实验(比如增进肌肉功能)中得到的。另一个值得一提的是中草药的安全使用历史经常在新成分申报中被引用,但是,除非可以证明历史上使用的中草药与申报中的膳食补充原料为同一成分并有着相同的使用剂量,FDA一般不会认为这样的历史使用数据可以作为唯一的安全依据。
结语
以上是对美国保健品市场的法规的一些浅析。保健品的标示和宣传同时受到FDA和美国联邦贸易委员会(Federal Trade Commission)的监管,近年来FDA也对保健品良好生产加工规范(cGMP)提出了更高的要求。对于中国保健品和原料的生产研发企业来说,了解和遵守这些繁复的法规是产品进入美国市场的一大挑战。尽管有些困难,但如果企业希冀在美国的保健品市场取得一席之地并获得成功,那就必须认真对待这些法规并把具体的法律调研作为产品商业计划的一部分。
专家介绍:
 陶鑫律师(Xin Tao)是Hogan Lovells华盛顿办公室的资深律师,其执业领域侧重于美国的食品药品法。凭借对美国监管系统的深刻了解以及自身的生物化学背景,陶鑫律师曾助力很多的食品、保健品及药品公司制定产品出口及上市的合规策划,并帮助企业应对政府调查。陶鑫律师同时参与主导了多个食品药品公司兼并收购的海外尽职调查。
陶鑫律师(Xin Tao)是Hogan Lovells华盛顿办公室的资深律师,其执业领域侧重于美国的食品药品法。凭借对美国监管系统的深刻了解以及自身的生物化学背景,陶鑫律师曾助力很多的食品、保健品及药品公司制定产品出口及上市的合规策划,并帮助企业应对政府调查。陶鑫律师同时参与主导了多个食品药品公司兼并收购的海外尽职调查。 Martin Hahn律师是霍金路伟律师事务所华盛顿办公室合伙人。Martin在食品及保健品领域具有丰富的执业经验,曾代理多家国际顶级食品以及保健品企业,以及为美国多个食品行业协会提供法律服务。Martin作为美国食品工业界律师的翘楚曾被权威的钱伯斯美国(Chambers USA)连续多年列为“第一流”(Band 1)。
Martin Hahn律师是霍金路伟律师事务所华盛顿办公室合伙人。Martin在食品及保健品领域具有丰富的执业经验,曾代理多家国际顶级食品以及保健品企业,以及为美国多个食品行业协会提供法律服务。Martin作为美国食品工业界律师的翘楚曾被权威的钱伯斯美国(Chambers USA)连续多年列为“第一流”(Band 1)。
相关热词搜索:
[责任编辑:]

 Mettler-Toledo 在中国国际渔业博览会上展示创新的产品
Mettler-Toledo 在中国国际渔业博览会上展示创新的产品
 食品异物问题频发?是时候了解X射线检测了
食品异物问题频发?是时候了解X射线检测了
 开拓科技创新,撬动橡塑业高质量发展
开拓科技创新,撬动橡塑业高质量发展
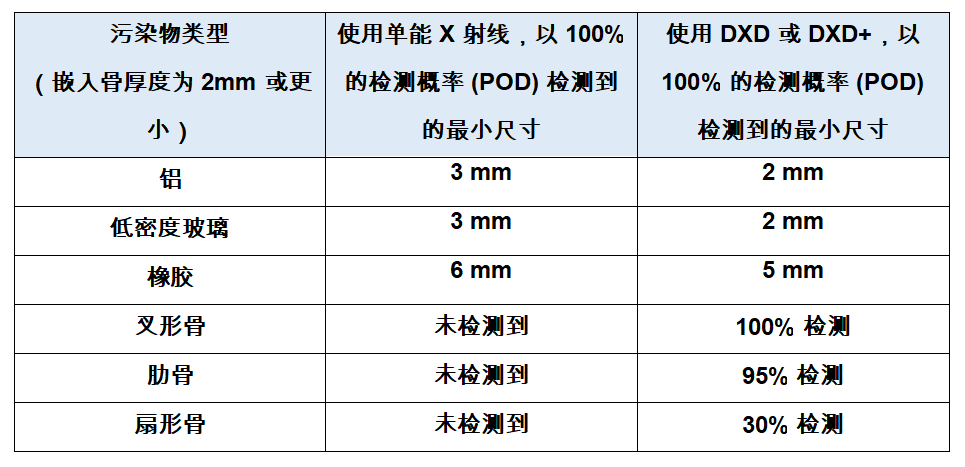 梅特勒托利多针对“难以发现”的污染物推出高品质X射线检
梅特勒托利多针对“难以发现”的污染物推出高品质X射线检
 探索婴幼儿辅食市场高质量发展之路,为宝宝成长保驾护航
探索婴幼儿辅食市场高质量发展之路,为宝宝成长保驾护航
 《食品安全最佳实践白皮书(2021-2022年)》四大主题发布
《食品安全最佳实践白皮书(2021-2022年)》四大主题发布
 《保健食品真实世界研究通则》团标技术审查与特食跨
《保健食品真实世界研究通则》团标技术审查与特食跨
 凝聚全球食饮智慧 SIAL西雅展国际化水平再创新高
凝聚全球食饮智慧 SIAL西雅展国际化水平再创新高
 精准把控 高质发展,第三届微生物安全与应用会议在
精准把控 高质发展,第三届微生物安全与应用会议在
 《食品行业科技创新白皮书》重磅发布!
《食品行业科技创新白皮书》重磅发布!






参与评论